在多相催化中,催化剂颗粒的尺寸和形貌会极大的影响催化活性,因此其结构-活性关系是多相催化研究中的核心问题之一。催化剂活性由表面电子结构决定,因而多数“结构-活性”研究都集中在构建“结构-能量”关系这一部分。从能量关系出发,通过求解微动力学模型,可以得到反应速率等动力学信息。对于多相催化中常见的纳米催化剂,目前最有效的微动力学模型是动力学蒙特卡洛模拟(KMC),但受制于计算量,其应用通常被局限于小粒径的纳米颗粒(<5nm)研究中。如果能够跨越DFT计算和KMC模拟步骤,直接构建催化剂表面活性位点的“结构-活性”关系,将大幅提升催化剂设计与筛选的效率,有力地扩展第一性原理在多相催化研究中的应用范围。
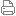
图1 a)构建“GCN-活性”关系示意图 b)三次多项式拟合纳米颗粒的“GCN-活性”关系 近日,我系徐昕教授课题组(https://xdft.fudan.edu.cn)发现,对于加氢/脱氢步骤控制的反应体系,由于氢在金属表面的吸附能对吸附位点不敏感,可以建立单个吸附位点与反应活性的对应关系,且纳米催化颗粒的活性可以由各表面位点的活性相加得到。基于单位点模型,徐昕教授课题组对Cu纳米颗粒催化的水汽变换反应进行了研究,并对大粒径Cu纳米颗粒(500-1000nm)的活性进行了预测,其预测表观活化能与实验值偏差小于1.0 kcal/mol。研究结果还表明,即使对于大粒径纳米颗粒,其催化活性也不能简单等价于面位的活性,例如对于877nm的正方体颗粒,其棱位对活性仍有较大贡献。这一研究成果以“Structure-Reactivity Relationship for Nano-Catalysts in the Hydrogenation /Dehydrogenation Controlled Reaction Systems”为题发表在《德国应用化学》杂志(Angew. Chem. Int. Ed. 2021, 60, 26342–26345)。课题组成员申同昊博士与博士生杨宇琦为共同一作。该工作得到了国家自然科学基金(Grant 21688102)及国家重点研发计划(Grant 2018YFA0208600)的支持。
全文链接:https://onlinelibrary.wiley.com/doi/10.1002/anie.202109942
文章来源:复旦大学
徐昕,复旦大学教授、博士生导师,国家杰出青年基金获得者、教育部长江学者特聘教授。1991年于厦门大学获得理论化学专业博士学位,1991年至1993年在福建物构所从事博士后研究,1993年开始任厦门大学化学系副教授,1995年任教授;1996年至2003年期间任厦门大学固体表面物理化学国家重点实验室副主任。2011年至今在复旦大学化学系工作。
| 







 发表于 2021-12-9 08:00:03
发表于 2021-12-9 08:00:03

 QQ好友和群
QQ好友和群 QQ空间
QQ空间 腾讯微博
腾讯微博 腾讯朋友
腾讯朋友 收藏
收藏 转播
转播 分享
分享 淘帖
淘帖